


Clin. Pract. 2024, 14(2), 619-628; https://doi.org/10.3390/clinpract14020049 (registering DOI) - 17 Apr 2024
Abstract
►
Show Figures
Penetrating aortic injuries represent critical medical emergencies that necessitate immediate intervention to prevent life-threatening consequences. When accompanied by the presence of an enormous right pleural false aneurysm, the clinical scenario becomes exceptionally rare and complex. This case report details the successful management of
[...] Read more.
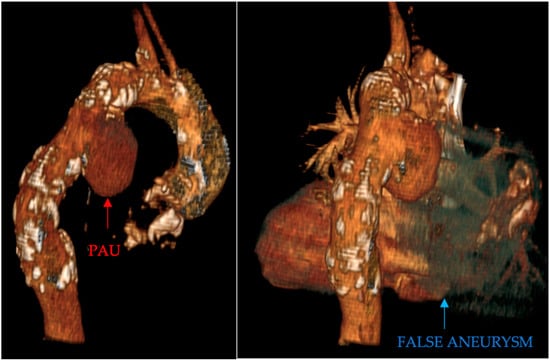
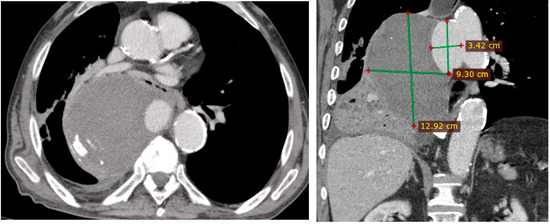
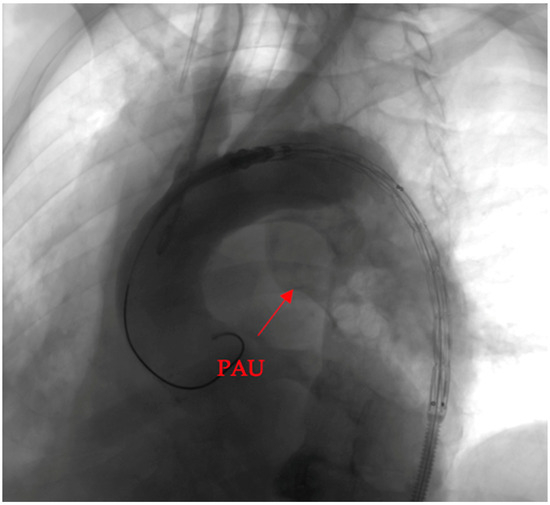
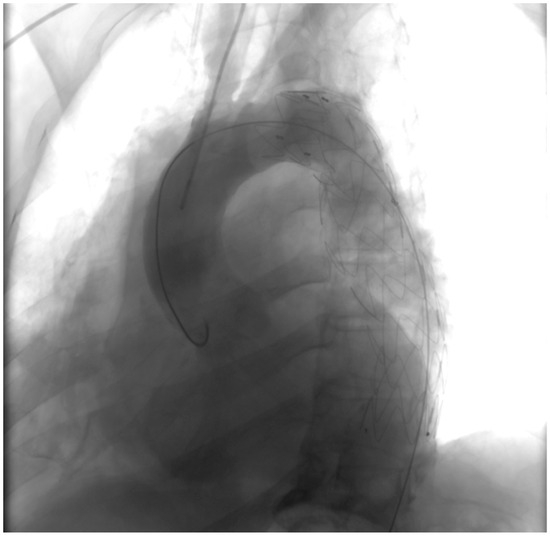
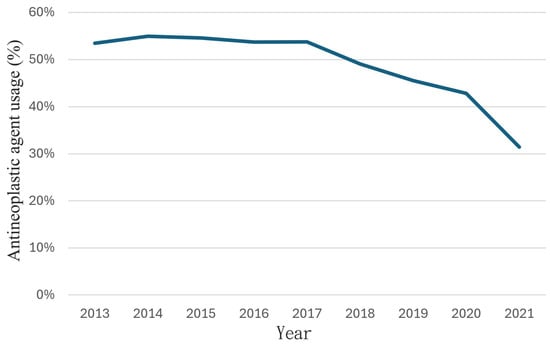
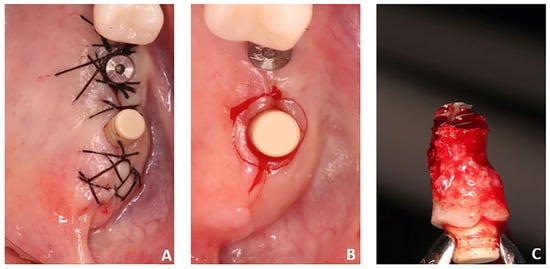
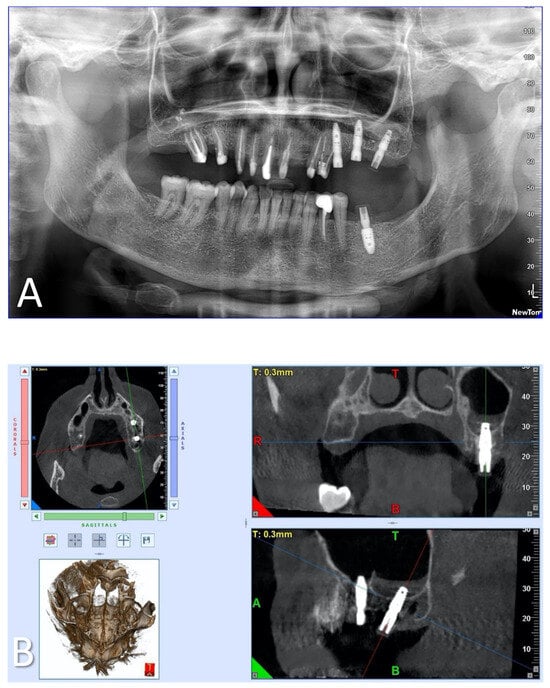
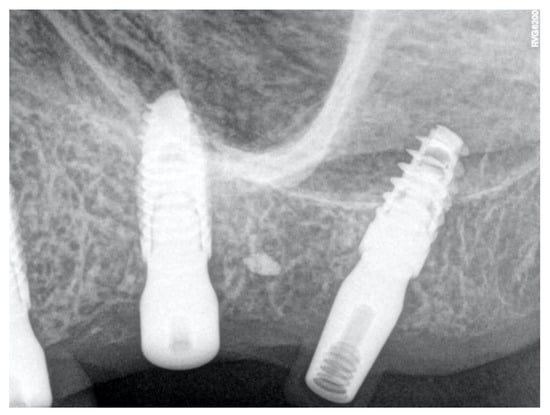
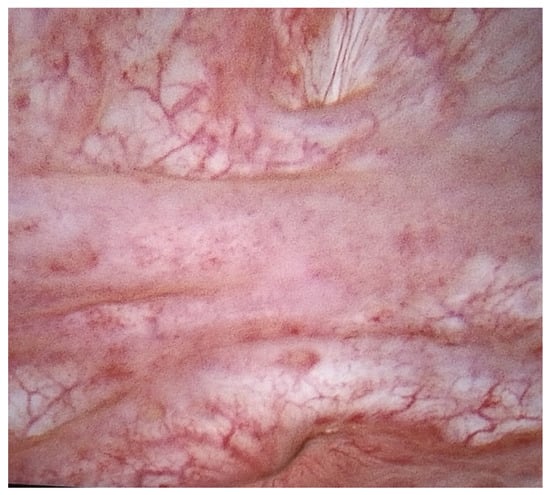
Penetrating aortic injuries represent critical medical emergencies that necessitate immediate intervention to prevent life-threatening consequences. When accompanied by the presence of an enormous right pleural false aneurysm, the clinical scenario becomes exceptionally rare and complex. This case report details the successful management of a patient who presented with a penetrating aortic ulcer and an extensive false aneurysm within the right pleura, employing an interdisciplinary approach involving cardiac surgeons, cardiologists, interventional cardiologists, and radiologists. The pivotal intervention involved the deployment of a covered and bare stent graft into the descending thoracic aorta to seal the aortic rupture. The patient’s clinical condition stabilized postoperatively, with no signs of recurrent hemorrhage. This case underscores the importance of rapid diagnosis, timely intervention, and the collaborative efforts of a specialized medical team in successfully managing such complex vascular injuries. Early recognition and referral to specialized centers are essential for improving patient outcomes in cases of penetrating aortic injuries with associated giant pseudoaneurysms.
Full article
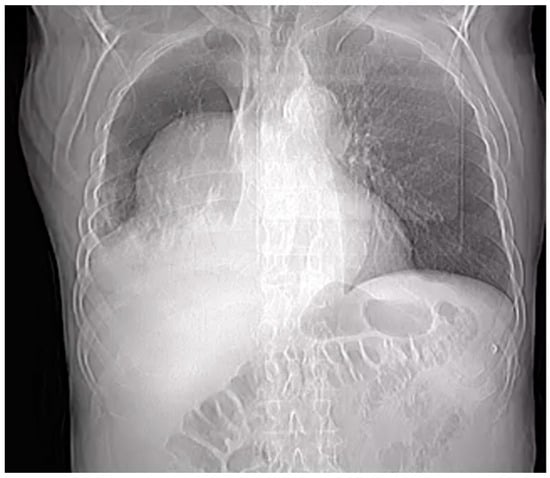
Figure 1



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}