


Clin. Pract. 2024, 14(2), 629-641; https://doi.org/10.3390/clinpract14020050 - 18 Apr 2024
Abstract
►
Show Figures
Purpose: We conducted a retrospective case series of seven male COVID-19 patients with respiratory failure and suspected OSA based on clinical features to evaluate the effects of undiagnosed obstructive sleep apnea (OSA) on COVID-19 outcomes and the response to a continuous positive airway
[...] Read more.
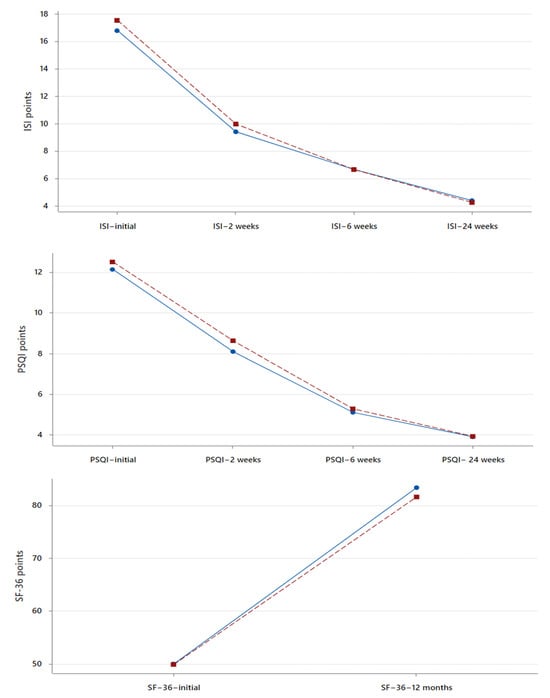
Purpose: We conducted a retrospective case series of seven male COVID-19 patients with respiratory failure and suspected OSA based on clinical features to evaluate the effects of undiagnosed obstructive sleep apnea (OSA) on COVID-19 outcomes and the response to a continuous positive airway pressure (CPAP) treatment. Cardiorespiratory polygraphy (CRP) and a continuous positive airway pressure treatment were used for diagnosis and management. They confirmed severe obstructive sleep apnea in all patients (apnea/hypopnea index > 30) and improved overnight oxygenation and symptoms at the 1-month follow-up. Conclusions: Undiagnosed obstructive sleep apnea may negatively impact COVID-19 outcomes by exacerbating respiratory failure. Recognition and treatment with continuous positive airway pressure can optimize the management of such patients.
Full article
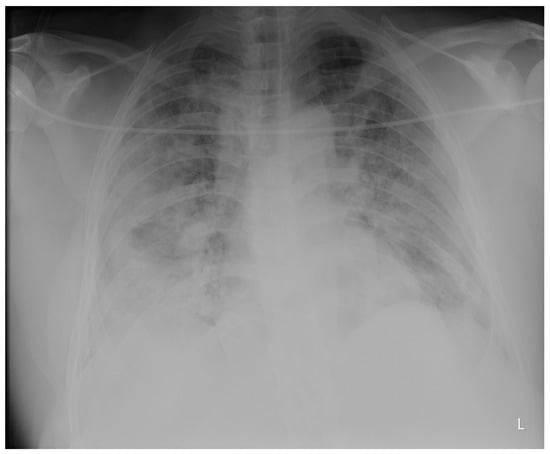
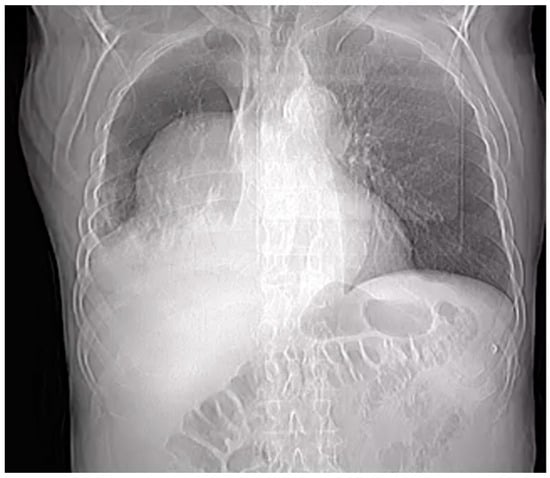
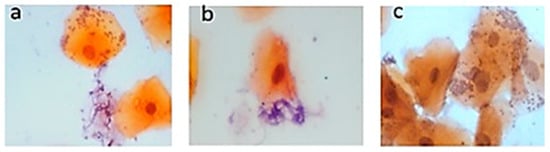
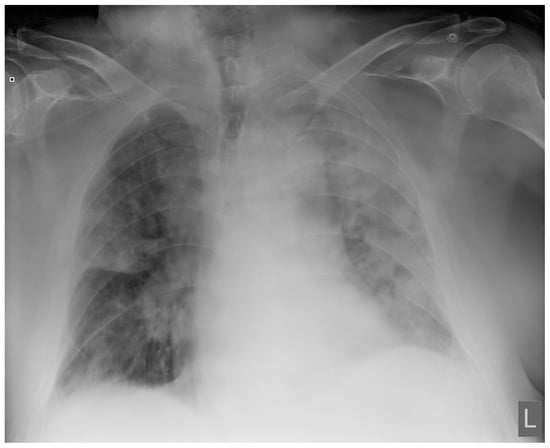
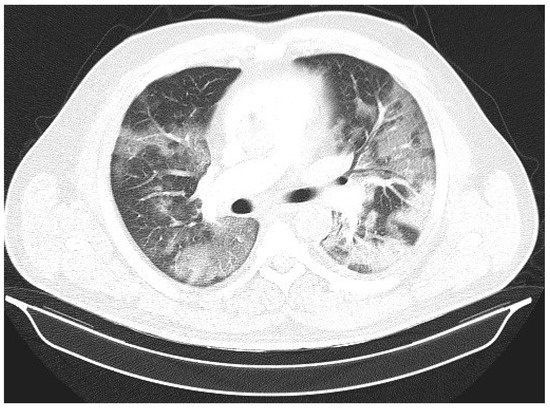
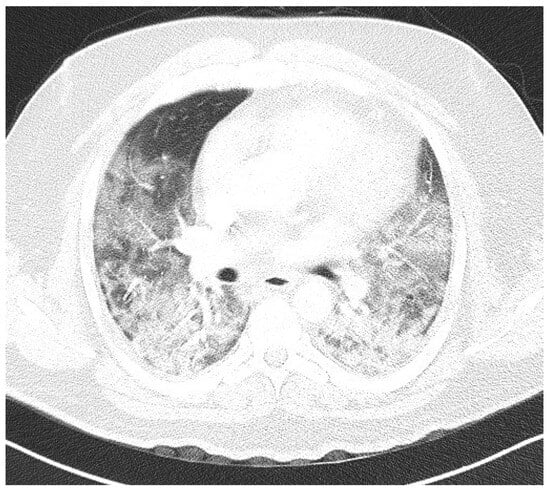
Figure 1



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}