


Clin. Pract. 2024, 14(3), 703-717; https://doi.org/10.3390/clinpract14030056 - 23 Apr 2024
Abstract
►
Show Figures
(1) Background: The aim of this study was to analyze the impact of pharmacogenetic-guided antidepressant therapy on the 12-month evolution of the intensity of depressive symptoms in patients with recurrent depressive disorder (RDD) in comparison to a control group of depressive subjects who
[...] Read more.
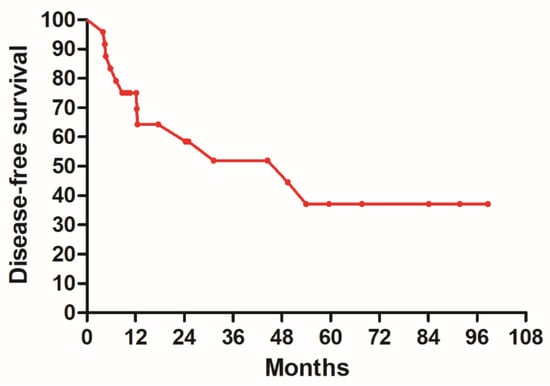
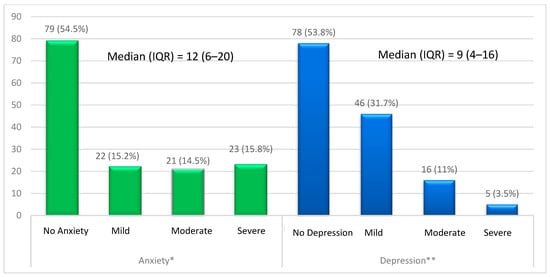
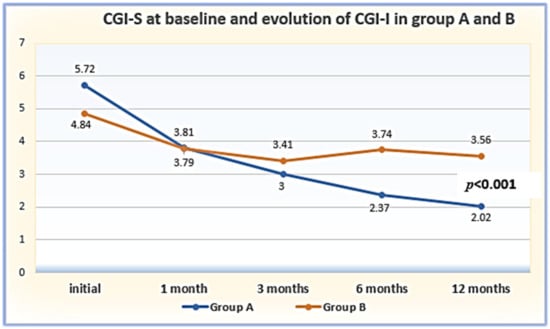
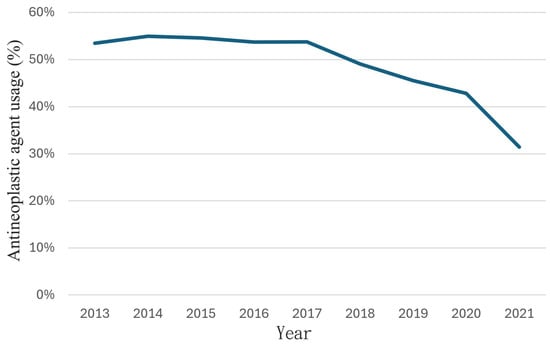
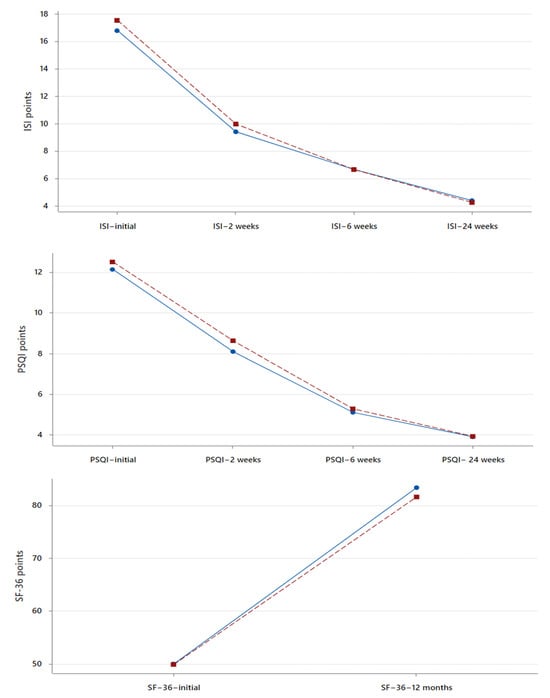
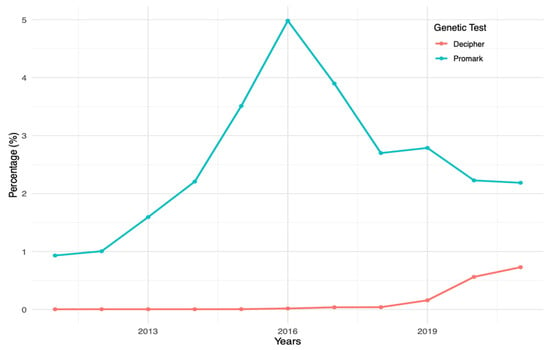
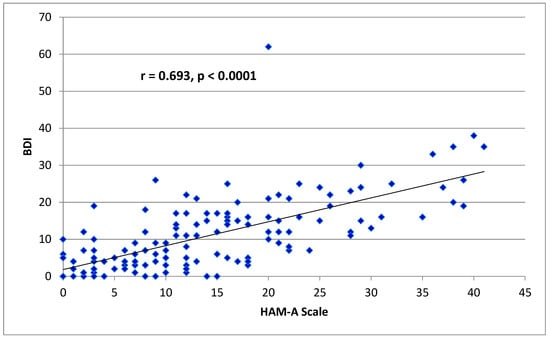
(1) Background: The aim of this study was to analyze the impact of pharmacogenetic-guided antidepressant therapy on the 12-month evolution of the intensity of depressive symptoms in patients with recurrent depressive disorder (RDD) in comparison to a control group of depressive subjects who were treated conventionally. (2) Methods: This prospective longitudinal study was conducted between 2019 and 2022, and the patients were evaluated by employing the Hamilton Depression Rating Scale (HAM-D), Hamilton Anxiety Rating Scale (HAM-A) and the Clinical Global Impressions Scale: Severity and Improvement. We followed them up at 1, 3, 6, and 12 months. (3) Results: Of the 76 patients with RDD, 37 were tested genetically (Group A) and 39 were not (Group B). Although the patients from Group A had statistically significantly more severe MDD at baseline than those from Group B (p < 0.001), by adjusting their therapy according to the genetic testing, they had a progressive and more substantial reduction in the severity of RDD symptoms [F = 74.334; η2 = 0.674; p < 0.001], indicating a substantial association with the results provided by the genetic testing (67.4%). (4) Conclusions: In patients with RDD and a poor response to antidepressant therapy, pharmacogenetic testing allows for treatment adjustment, resulting in a constant and superior reduction in the intensity of depression and anxiety symptoms.
Full article
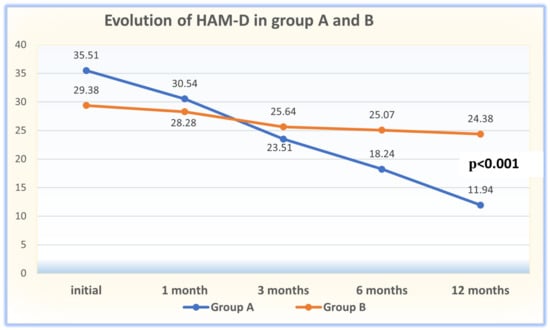
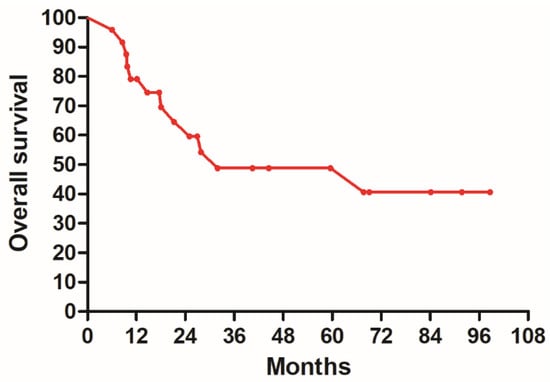
Figure 1



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}