


Clin. Pract. 2024, 14(3), 739-748; https://doi.org/10.3390/clinpract14030059 (registering DOI) - 25 Apr 2024
Abstract
►
Show Figures
Introduction: Intra-abdominal cystic formations represent heterogeneous pathologies with varied localization and clinical manifestation. The first challenge of a giant intra-abdominal cystic lesion is identifying the organ of origin. The clinical presentation of intra-abdominal cystic lesions varies from acute manifestations to non-specific symptoms
[...] Read more.
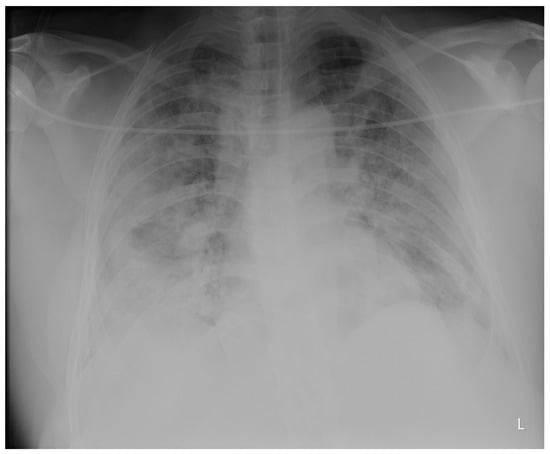
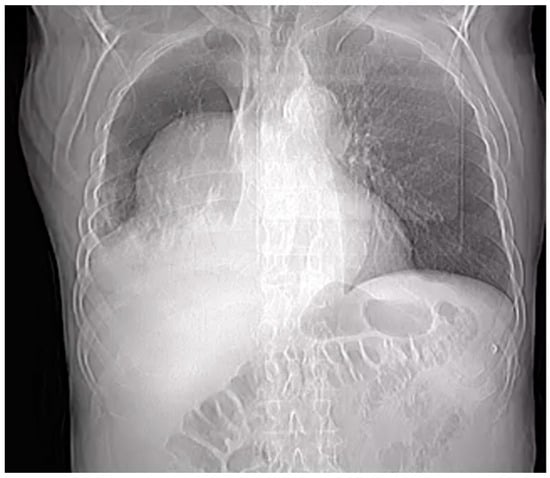
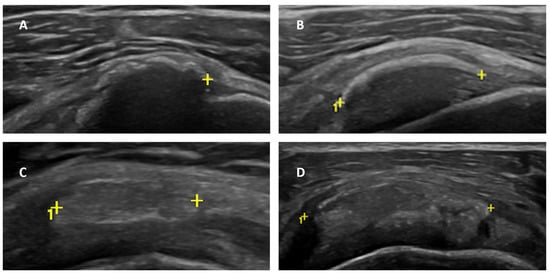
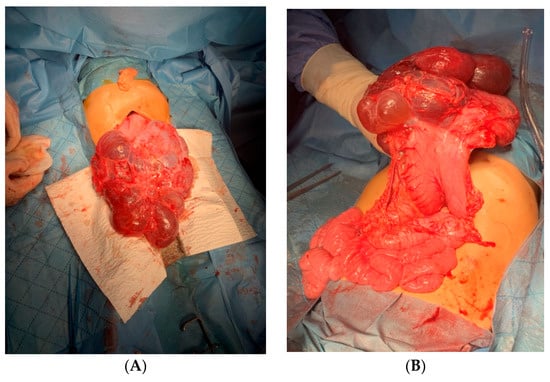
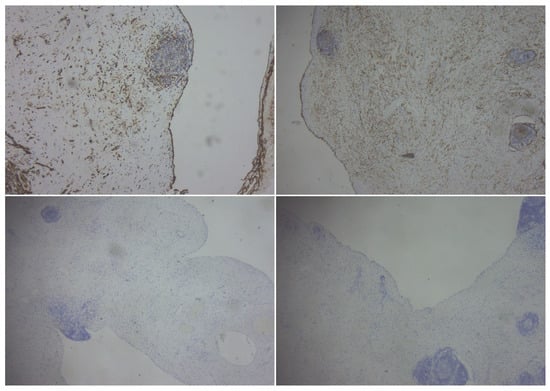
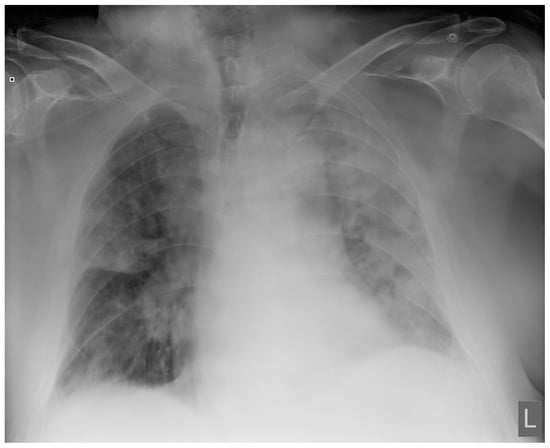
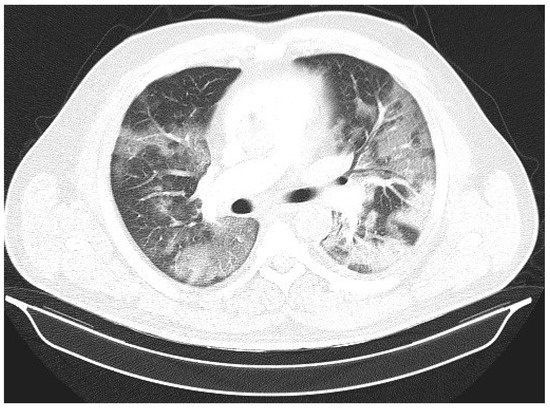
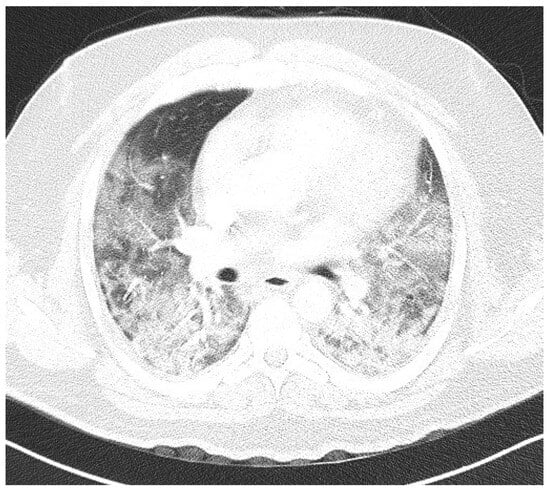
Introduction: Intra-abdominal cystic formations represent heterogeneous pathologies with varied localization and clinical manifestation. The first challenge of a giant intra-abdominal cystic lesion is identifying the organ of origin. The clinical presentation of intra-abdominal cystic lesions varies from acute manifestations to non-specific symptoms or accidental discovery. Case presentation: A 2-year-old girl presents to the emergency unit with a fever of 38.5 Celsius, loss of appetite, and apathy. The investigations showed a gigantic intra-abdominal mass whose organ belonging could not be specified. Postoperatively, a giant mesenteric lymphangioma was evident, which was completely excised. Discussion: Giant cystic formations modify the anatomical reports and become space-replacing formations, and the starting point is even more challenging to assess preoperatively. Nevertheless, the careful evaluation of the characteristics of the formation, the effect on the adjacent organs, the age of the patient, and the clinical picture can provide elements of differential diagnosis. The stated purpose of this work is to systematize intra-abdominal lesions according to the organ of origin and to make the preoperative diagnosis of an intra-abdominal cystic lesion in the pediatric patient easy to perform starting from the presented case.
Full article
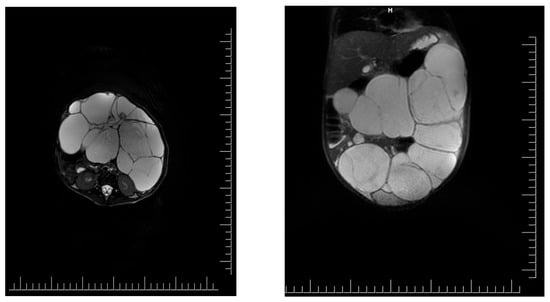
Figure 1



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}